|
|

|

|
| e-Pub |
Section: Research Program
RNA and protein structures
At the secondary structure level, we contributed novel generic techniques applicable to dynamic programming and statistical sampling, and applied them to design novel efficient algorithms for probing the conformational space. Another originality of our approach is that we cover a wide range of scales for RNA structure representation. For each scale (atomic, sequence, secondary and tertiary structure...) cutting-edge algorithmic strategies and accurate and efficient tools have been developed or are under development. This offers a new view on the complexity of RNA structure and function that will certainly provide valuable insights for biological studies.
Discrete representations and complexity
Participants : Yann Ponty, Wei Wang, Antoine Soulé, Juraj Michalik.
Common activity with J. Waldispühl (McGill) and A. Denise (Lri ).
Ever since the seminal work of Zuker and Stiegler, the field of RNA bioinformatics has been characterized by a strong emphasis on the secondary structure. This discrete abstraction of the 3D conformation of RNA has paved the way for a development of quantitative approaches in RNA computational biology, revealing unexpected connections between combinatorics and molecular biology. Using our strong background in enumerative combinatorics, we propose generic and efficient algorithms, both for sampling and counting structures using dynamic programming. These general techniques have been applied to study the sequence-structure relationship [46], the correction of pyrosequencing errors [38], and the efficient detection of multi-stable RNAs (riboswitches) [42], [43].
Increasingly, we develop and study parameterized complexity approaches, based on dynamic programming over a tree decomposition, for several combinatorial problems, including RNA design, structure-sequence alignment (aka threading in the context of proteins). The later problem is at the core of Wei Wang's Phd, successfully defended in Dec 2017. In the context of our probabilistic approaches, often based on random generation, such parameterized algorithms usually follow proofs of hardness for the associated enumeration problems.
|
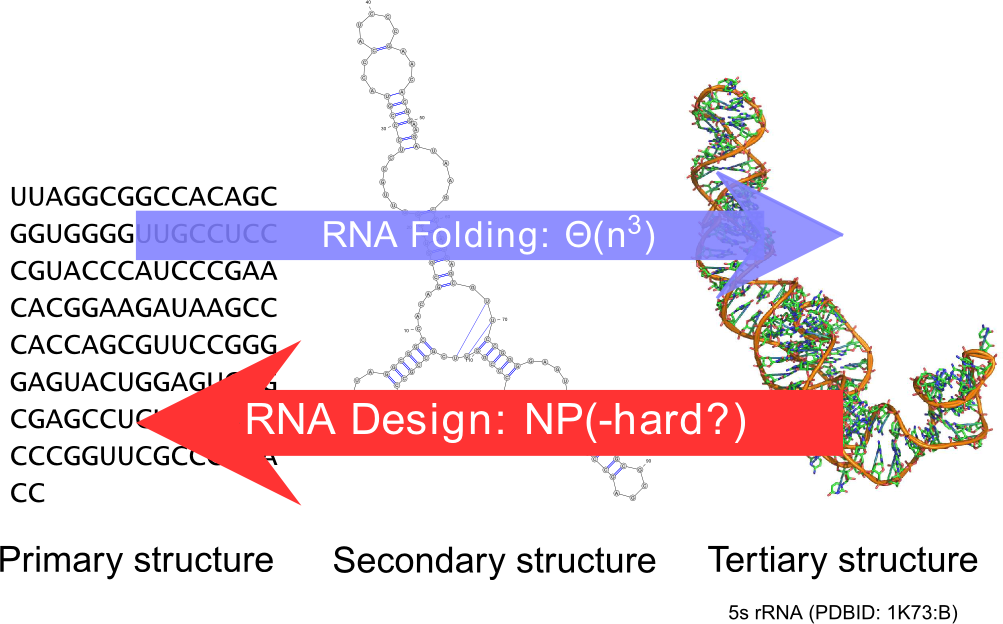
RNA design.
Participants : Alice Héliou, Yann Ponty.
Joint project with A. Denise (sc Lri), J. Waldispühl (McGill), D. Barash (Univ. Ben-Gurion), and C. Chauve (Simon Fraser University).
It is a natural pursuit to build on our understanding of the secondary structure to construct artificial RNAs performing predetermined functions, ultimately targeting therapeutic and synthetic biology applications. Towards this goal, a key element is the design of RNA sequences that fold into a predetermined secondary structure, according to established energy models (inverse-folding problem). Quite surprisingly, and despite two decades of studies of the problem, the computational complexity of the inverse-folding problem is currently unknown.
Within AMIBio, we develop a new methodology, based on weighted random generation [25] and multidimensional Boltzmann sampling, for this problem. Initially lifting the constraint of folding back into the target structure, we explored the random generation of sequences that are compatible with the target, using a probability distribution which favors exponentially sequences of high affinity towards the target. A simple posterior rejection step selects sequences that effectively fold back into the latter, resulting in a global sampling pipeline that showed comparable performances to its competitors based on local search [32].
The main advantages of this approach is its linear complexity, and its flexibility in incorporating constraints. Indeed, extensive experiments revealed a drift of existing software towards sequences of high G+C -content, and we showed how to control this distributional bias by using multidimensional Boltzmann sampling [37], [36]. Recently, we are extending this approach to the design of RNAs with multiple structures, developping a Fixed-Parameter Tractable framework that naturally extends to capture negative design goals.
Modeling large macromolecular architectures
Participants : Yann Ponty, Afaf Saaidi, Mireille Régnier, Amélie Héliou.
Joint projects with A. Denise (Lri) , D. Barth (Versailles), J. Cohen (Paris-Sud), B. Sargueil (Paris V) and Jérome Waldispühl (McGill).
The modeling of large RNA 3D structures, that is predicting the three-dimensional structure of a given RNA sequence, relies on two complementary approaches. The approach by homology is used when the structure of a sequence homologous to the sequence of interest has already been resolved experimentally. The main problem then is to calculate an alignment between the known structure and the sequence. The ab initio approach is required when no homologous structure is known for the sequence of interest (or for some parts of it). We contribute methods inspired by both of these directions.
We also develop homology-based approaches for structure modeling, and developed a general setting for the problem of RNA structure-sequence alignment, known to be NP-hard in the presence of complex topological features named pseudoknots (PKs). Our approach is based on tree decomposition of structures and gives rises to a general parameterized algorithm, where the exponential part of the complexity depends on the family of structures [39]. This work unifies and generalizes a number of recent works on specific families, and enables the curation of multiple alignments for RNA families featuring PKs, correcting certain bias introduced by PK-oblivious methods.